
随着药物研发全球化,国际多中心临床试验(multi-regional clinical trial,MRCT)日益受到重视。MRCT的实施可以加快新药同步研发,使得当试验结果用于多个监管机构注册审评时,维持试验设计在相同水平的科学严谨性;同时可以优化宝贵的患者资源和减少不必要的研发费用。但MRCT也给各国药品监管带来了挑战,不少国家,包括中国和国际组织如ICH都出台过相关的规定。本文对比了美国、欧盟和日本的MRCT监管要求,介绍了ICH相关指导原则及其与中国监管要求的异同,以期对我国MRCT的监管工作有所启发。
6193字 | 9分钟阅读
Key words:Multi-regional clinical trial/ICH/Regulatory requirements
随着药物研发全球化,新药临床研发模式发生了根本改变,主要地区从原来的美国、欧洲、澳大利亚,扩展到同时在美洲、欧洲(包括东欧)、亚洲、拉丁美洲等国家和地区开展。国际多中心临床试验(multi-regional clinical trial,MRCT)是在同一个方案下多个地区(这里的地区包含地理区域、国家或者监管区域)进行的临床试验。因MRCT可以维持试验设计在相同水平的科学严谨性,减少不必要 的临床试验重复,获取全球的临床数据,为每一个地区药物批准提供更好的证据,缩短药品研发上市时间,在计划于多个地区申报注册的药物研究中,MRCT愈受青睐。
1、各国MRCT监管要求对比
MRCT的数据能否完全适用于不同国家或地区的患者人群,受到诸多内在和外在因素的影响,成为各国药监部门审评审批的一大挑战。针对这个问题,不少国家都曾出台过一些本国的规定。如日本 PMDA在2007年9月就提出过《国际临床试验基本原则》(Basic principles on Global Clinical Trials),对如何接受日本人以外的数据予以澄清解释;EMEA在2009年发表《欧洲以外临床试验结果外推至欧洲人群的考虑要点》(Reflection paper on the extrapolation of results from clinical studies conducted outside of the EU to the EU population),分析了研究结果外推至欧洲人群可能出现的差异及需要关注的问题。其研究结果显示,在外在因素中,例如医疗实践、疾病定义及研究人群可能对国外研究数据外推至欧洲人群的适用性造成影响。美国《FDA 的观点: 关于用于支持在美国的新药申请中使用外国人数据的监管和科学问题》(regulatory and scientific issues regarding use of foreign data in support of new drug applications in the United States:an FDA perspective) 一文中,FDA在监管中发现了MRCT的很多问题,比如越来越多的试验使用国外数据,患者标准治疗和实际用药剂量不一样,等等。最后得出结论认为,MRCT数据要被美国接受,统一标准以及与各方紧密合作是关键。本文对比分析了美国、欧盟和日本对MRCT监管的要求,见表1。

2、ICH相关指导原则
ICH(International Conference on Harmonization,人用药品注册技术规范国际协调会,2015年更名为International Council for Harmonization)是欧共体、美国和日本在1990年发起成立的,对三方成员国家的人用药品注册技术要求的现存差异进行协调的国际组织,一直致力于起草和发展能符合会员国之间法规基本要求的统一准则和药物开发及注册标准。
在过去20多年里,ICH发布了一系列质量体系(Q1~Q12)、安全性(S1~S11)、有效性(E1~E18)和多学科(M1~M10)指南,促使研究和申报符合同等标准,以期缩短药品研发上市时间,帮助患者获得经济实惠的药品。
其中,ICHE是与人类临床研究相关的课题,关注临床试验的设计、实施、安全性和报告等。同时还涵盖了一些利用生物技术及药物基因组学技术得到有更好靶向性的新物。
2.1 ICH E5
ICH E5《接受国外临床试验数据中有关种族因素的指导原则》(R1)及“E5指南问答”一直是新药全球临床研发最基本和最重要的指导文件之一。ICH E5是由日本于1992年向ICH管理委员会提议立题的,1998年2月进入步骤4,1998年3月ICH E5(R1)批准发布。其目的是通过推荐一个用于评估种族因素对药物作用影响的框架,如某一剂量和给药方案对某药的安全性和有效性的影响,促进药物在ICH地区的注册。它描述了:
1) 便于外推到不同人群,能够支持在新地区注册的国外临床试验数据的特征。
2) 减少临床试验重复,加速新地区上市批准的监管策略。
3) 如何使用桥接试验,将国外临床试验数据外推到新地区。
4) 能够描绘出种族因素对安全性、有效性、剂量和给药方案的影响的研发策略。
ICH E5提出了2个重要的概念。一个是桥接试验,定义为:为了提供新地区的药效学和/或有效性、安全性、剂量和给药方案的临床数据,使得国外临床数据能够外推到新地区人群,而在新地区进行的试验。另一个是种族因素,被定义为与人群的遗传基因和生理病理相关的,能帮助判断与鉴别亚人群,可能影响临床数据在地区间外推的因素(内在的),以及与居住地环境和文化(外在的) 特征有关 的因素,具体因素与分类见表2。
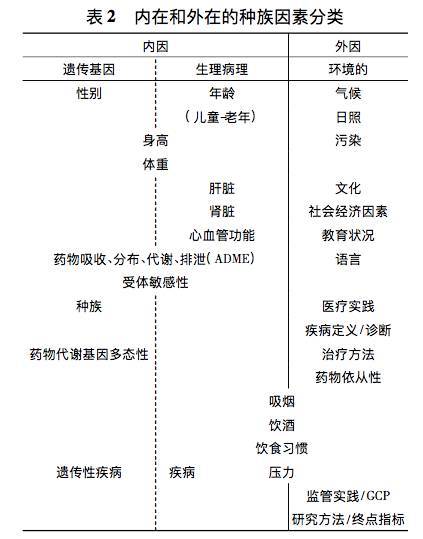
ICH E5为药物监管和研发策略提供了指南,在尽可能减少临床试验重复和尽快为患者提供药物的同时,对种族因素的影响进行充分的评估,有效减少了新地区申请新药注册的工作量,缩短了药品在新地区的上市时间。
2003年,ICH“E5指南问答”发布,澄清了最初指南中不清晰和容易导致误解的地方。2006年,“E5指南问答(R1)”发布,在其Q&A10中提到,随着地区间相互接受国外临床试验数据经验的累积,将会进一步了解桥接试验的适用情况。随着这些经验的增加,桥接试验的需求将会减少。而在Q&A11中,进一步为桥接引入了国际多中心临床试验的概念,并首次提出MRCT作为进行桥接的策略。
2.2 ICH E17
在ICH E5及“E5指南问答”发布后,业内已经累积了十几年的MRCT的经验。ICH意识到可以将这些经验整合成为新的指南,从而推进没有单独桥接试验的MRCT的使用。于是,ICH在2014年6月启动了ICH E17的制定工作。到2016年6月,专家工作小组已经完成了一致性草稿,目前正处于步骤3。从2016年7月起,到2017年1月止,ICH E17分别在日本、美国、欧盟开放公众咨询,收集公众的意见与建议,并分别于2016年11月与2017年6月进行第4次和第5次面对面工作会,根据公众咨询中收到的意见与建议对指南草稿进行修改。ICH计划于2017年11月完成步骤4,最终定稿,并于第四季度开始E17指南的应用。ICH E17将补充ICH E5及“E5指南问答”的不足,提高多国/地区监管部门对MRCT数据的接受程度。在此,我们尝试将ICH E17一致性草稿中重要的信息翻译整理出来,以供大家参考和学习。
ICH E17制定的目标是为了提高MRCT在全球监管部门的可接受性,描述统一规划与设计MRCT时的基本原则。这份指南并不是独立的,应与其他E类指南一同使用。
当计划在多个地区同时提交药品上市申请时,MRCT往往是最优选择。开展MRCT的前提假设是药品治疗效果在所有相关地区有临床意义,但同时需认识到这些地区间也会存在疗效差异。在计划MRCT时,种族因素是要考虑的关键点。这些因素应当在计划时期就做好定义,在MRCT实施阶段,应该收集并评估所有与之相关的信息。

2.3 ICH E17重点关注-MRCT规划与设计中重点考虑的问题
MRCT使在不同人群中检验治疗效果成为可能,可以加快新药同步研发,使得当试验结果用于多个监管机构注册审评时,维持试验设计在相同水平的科学严谨性,并且通过在多个地区同时进行临床试验,减少各个地区单独进行临床试验的次数,提高全球药物研发的效率。作为新药全球研发过程中临床试验的一种新的组织形式,相比一般的多中心临床试验,MRCT在设计、管理、实施、分析等方面也更加复杂。在最新的E17草稿中,提出了MRCT规划与设计中需要考虑的一些问题。
2.3.1 策略相关问题
进行MRCT最基本的要求是所有参与地区应共享同一个研究假设,使用相同 的对照药及在所有地区都具有临床意义的主要终点。此外,如果预测到在药物反应上存在种族差异, 那么应当在规划和设计验证性MRCT前进行探索性MRCT,以检验这些差异的大小,并进行更多的药动学试验来进一步了解种族差异。在规划阶段,应仔细考虑地区间差异以及此差异可归因于内在和外在因素的程度。同时,理解相关监管地区不同监管要求也是十分重要的,最好在规划阶段就能针对MRCT的整体方案和数据的可接受性与监管机构进行沟通。在全球水平上申办方与监管机构之间的有效沟通可以促进药物研发的进程。
2.3.2 试验设计及方案问题
在MRCT的试验设 计和方案中需要注意并理解以下问题:
1) 地区变异性及其对疗效和安全性的影响
在MRCT规划阶段,应充分考虑地区变异性及其可用内外在因素解释的程度。尽可能地搜索最新及最相关的数据,用 来解释地区变异性的可能原因。如果使用历史数据,需要从科学和方法学上考虑这些数据是否依然相关,是否与现在的治疗环境相符。
2) 主要终点及次要终点
主要终点必须具有临床意义,能够被医学实践所接受,具有足够的敏感性和特异性,以便于发现预期的治疗效果。主要终点及次要终点必须能被所有相关监管机构接受,以确保MRCT成功与否的解释在各地区和监管机构中是一致的。同时,尽可能保持地区间次要终点的统一,以提高试验可行性和质量。
3) 整体样本量和地区样本量分配
确定总体样本量的基本原则是所有地区样本量总和在统计学意义上足以验证主要的研究假设。而分配各地区样本量时,应主要基于有效性而非安全性,能够合理地描述和评估地区间有临床意义的治疗效果的差异,且不会大幅增加基于主要研究假设得出的样本量。分配样本量的方法都有一定的局限性,目前无统一可接受的或者标准化的方法可用。应综合考虑地区的大小、疾病流行模式、各地区基于内在和外在因素招募受试者的共性、后勤保障等,采用一个平衡的办法,以确保试验可行,并能为各地区的药物评价提供足够的信息。
4) 合并地区和/或合并亚组
进行MRCT时,当某些地区的个体在内外在因素上相似时,可将这些地区合并为一个地区,即为合并地区,如东亚、欧洲和北美等。同时也可以将一个特定 的地区中的受试者的一个子集与其他地区的类似定义的子集合并,形成一个合并亚组。其成员共享一个或多个内在或外在的对药物研发重要的因素。如生活在北美和南美的西班牙裔。要注意的是,合并地区或亚组需要在试验研究规划阶段就具体说明并在研究方案中描述,作为相关监管机构做出监管决策的基础。
5) 统计分析计划
进行统计分析计划时应注意几个方面:
首先,在试验开始前同监管机构讨论确定主要分析方法。如果因为监管或者科学的原因,不同监管机构要求不同的分析方法时,在试验方案中要一一描述出来。
第二,建议在设计阶段计划亚组分析,并在方案以及统计分析计划中预先定义好。对内外在因素影响的分析进行合乎情理、前瞻性的计划,可以尽可能地减少数据驱动的事后亚组分析,为区域一致性评价建立较好的基础。同时,应该提前计划如何分析和评估治疗效果,以便定性 / 定量的评估亚组和各地区的获益风险比。
第三, 在主要疗效分析中考虑地区因素。如果MRCT是按照地区进行随机的,那么在进行主要疗效分析时,需要应用合适的统计方法来调整地区的影响。
第四,统计分析计划应当包括地区间治疗效果一致性的评价方法,以及在地区间观察到的有意义的临床差异应当如何用内在和/或外在因素来解释。
第五,如果样本量足够的话,统计分析计划应该描述每一个地区治疗效果评估与报告的统计方法及指标测量方法,其分析策略应与主要分析策略一样。
最后是MRCT实施的监查稽查与减少地区间的试验质量差异。
结合ICH E9中关于试验设计统计原则的一些基本考虑,ICH E17指南将使MRCT的规划、设计及管理更加完善,提升药物研发的科学严谨性及效率,避免药物研发过程中重复的工作,得到更好的监管决策。这份指南将为MRCT的规划与设计提供一个统一的方法,使监管机构间矛盾的观点最小化,从而更好地接受并充分利用在多个地区进行MRCT得到的数据。

3、中国MRCT指南与ICH E17的异同比较
为指导和规范MRCT在中国实施与开展,CFDA于2015年1月30日发布了《国际多中心药物临床试验指南(试行)》,自2015年3月1日起试行。我们对比分析了中国MRCT指南与ICH E17的异同,见表3。可以看出,中国指南和ICH E17的基本概念和核心信息是一致的,在充分体现与国际接轨的同时,更强调了国家的概念,并增加了中国药监的一些特别关注。

指南发布后,国内各相关机构和协会针对MRCT的设计、实施和监管组织了多次讨论,尤其在科学性方面,包括种族差异、亚组界定及所占比例、对照药的选择、一致性评价的标准、差异评价等均有所产出。如2015年6月至10月,北京大学监管科学卓越中心和哈佛布莱根妇女医院MRCT中心针对MRCT内源性和外源性因素对评价药物安全性和有效性的影响、怎样定义MRCT的区域、亚组一致性评价和样本量的考虑等进行了为期4个月的讨论,并联合举办了“ICH E17科学监管研讨会”,进一步研讨和汇报了研究的科学成果和政策建议。
MRCT一致性工作组提出了3个不同级别的疾病分类及相应的一致性定义(从弱一致性到强一致性):根据不同的疾病种类,可以考虑相应的3个不同级别的“一致性”。
级别1:亚组结果与整体结果呈相同的趋势;
级别2:亚组结果与整体结果( 疗效) 呈一定比例;
级别3:亚组结果与整体结果同时具有临床显著性和统计严谨性。
根据不同一致性的定义,在考虑MRCT的区域、亚组结果的一致性的时候,使得评价更趋于量化和精确。
同年7月,中国学者发表了“探讨药物国际多中心临床试验设计的一种新方法SGDDP”。SG-DDP(Simultaneous Global Drug Development Pro-gram)设计的总体思路是,除用于原来区域注册的MRCT外,根据需要可以考虑纳入一个当地试验(LCT)试验,并且LCT和MRCT具有相同的设计,以此来支持在当地国家(新区域)的注册。SGDDP中的MRCT是一个标准的III期试验的设计,预先设定治疗效应大小和统计学显著水平。LCT则可以视为MRCT的一个延展。MRCT中入选有目标种族人群(TE)和非目标种族人群(NTE)患者,而LCT入选的均为TE患者。通过采用加权的Z-统计量来合并MRCT中收集到的信息和LCT收集到的信息,降低非目标种族人群在加权检验统计量中的信息权重,并严格控制统计假设的I类错误,从而检验评估在目标种族人群(TE)的有效性。
MRCT在我国尚属新兴尝试,其规则也处于不 断地探索和制定过程,不同于以往的翻译和转化,需要国家食品药品监督管理总局(CFDA)的持续参与,对CFDA也是一种尝试和挑战。相信随着业内的广泛讨论和MRCT的持续开展,相关指南会得到进一步的细化和完善。
通过以上对比,我们发现各国/地区对MRCT监管要求的基本理念都是相似的,只是在具体要求上存在一些差别,这跟各国/地区的药物研发和监管现状以及医疗实践差异等都是相关的。我们建议进 一步加强国际监管合作和交流,以便更好地理解和使用国际通用标准,推动中国尽快加入药物研发的全球化进程,缩短创新药在中国上市的时间差,从而真正实现以患者为核心的药物研发和监管。
2017年3月17日,CFDA起草了《国家食品药 品监督管理总局关于调整进口药品注册管理有关事项的决定(征求意见稿)》,并向社会公开征求意见。该征求意见稿提出:
1) 在中国进行MRCT者,取消临床试验用药物应当已在境外注册或者已进入 II期或者III期临床试验的要求,疫苗类药物除外。
2) 对于在中国进行的MRCT,完成MRCT后,可以直接提出药品上市注册申请; 提出上市注册申请时, 应当执行《药品注册管理办法》及相关文件的要求。
3) 对于申请进口的化学药品新药以及治疗用生物制品创新药,取消应当获得境外制药厂商所在生产国家或者地区的上市许可的要求。
4) 对于本决定发布前已受理的,以MRCT数据提出免做进口临床试验的注册申请,符合要求的,可以批准进口。
此征求意见稿的发布表明了我国鼓励和推动MRCT的努力和决心,开启了我国监管理念革新的新篇章,将为优化中国创新药物研发生态环境,提高中国创新药物研发能力,帮助公众尽快的用到好药提供积极的政策引导和坚实的政策支持。相信随着中国药监改革的深化和推进,在一系列MRCT相关指南的引导下,有中国参与的MRCT全球同步研发及上市指日可待。
(志谢:感谢美国默克研究实验室生物统计和科学决策部执行总监王武保博士、北京大学临床研究所武阳丰教授对本文撰写与修改提出的宝贵建议与意见。)

声明:该文系转载,不以任何商业为目的,仅供参考和学习交流,如有违背著作方权益和利益,可立即删除。